MurciaSalud
CSUR ERN
¿Qué necesito saber acerca del Síndrome de QT Largo?
El síndrome de QT largo (SQTL) se caracteriza por una anomalía de la actividad eléctrica cardíaca que se puede diagnosticar realizando un registro de los impulsos eléctricos del corazón (electrocardiograma o ECG). El corazón a pesar de tener una anatomía normal presenta una disfunción eléctrica (intervalo QT anormalmente largo).
El síndrome está asociado a un riesgo elevado de síncopes (desmayos), pérdida de conciencia, alteraciones del ritmo cardiaco, e incluso paradas cardíacas, que pueden producirse por realizar esfuerzos o por la administración de ciertos medicamentos. (ver anexo de la ficha Orphanet Urgencias sobre el Síndrome de QT largo).
El síndrome de QT largo es con frecuencia hereditario (familiar), de origen genético y habitualmente congénito (presente al nacimiento).
Existen varias formas específicas del síndrome de QT largo:
- el síndrome de Romano-Ward es la forma más frecuente (forma autosómica dominante);
- el síndrome de Jervell y Lange-Nielsen, muy poco frecuente, asociado a sordera (forma autosómica recesiva);
- el síndrome de Timothy, excepcional, asociado a malformaciones de las manos y de la cara, y alteraciones del desarrollo cercanas al autismo (forma autosómica dominante).
¿Está presente en toda España y el mundo? ¿Cuántas personas están afectadas?
El síndrome de QT largo está presente en todo el mundo pero su frecuencia varía de una región a otra. En Europa, el número de casos al nacimiento por año (prevalencia al nacimiento) se estima en 1 por cada 2.000 nacimientos. En la mayoría de casos, se trata del síndrome de Romano-Ward. La prevalencia estimada del síndrome de Jervell y Lange-Nielsen es de 1 caso por 200.000 a 1 caso por 1.000.000.
Las complicaciones del síndrome de QT largo familiar se producen sobre todo en personas jóvenes de entre 10 y 30 años, y con menor frecuencia en mujeres que en hombres.
¿Cuál es la causa?
El síndrome de QT largo familiar es de origen genético. Es debido a la alteración (mutación) de un gen. Los genes son trozos de ADN, sustancia que constituye los cromosomas y que contiene nuestro patrimonio genético. Un gen equivale a un "manual de instrucciones" que da las órdenes para producir una proteína. Las proteínas tienen funciones muy variadas: contribuyen al funcionamiento normal de cada célula, y más globalmente, del organismo. Hasta la fecha han sido identificados dieciséis genes cuyas mutaciones causan un síndrome de QT largo familiar, entre los que se encuentran: KCNQ1 (da lugar al síndrome de QT largo tipo 1 o SQTL1), KCNH2 (síndrome de QT largo tipo 2 o SQTL2), KCNE1, KCNE2, SCN5A (Síndrome de QT largo tipo 3 o SQTL3), SCN4B, ANK2, CAV3, AKAP9, SNTA1, ALG10, KCNJ5, NOS1AP, CACNA1C. Todos estos genes codifican proteínas necesarias para el control de la actividad eléctrica cardíaca (ver figura 1).
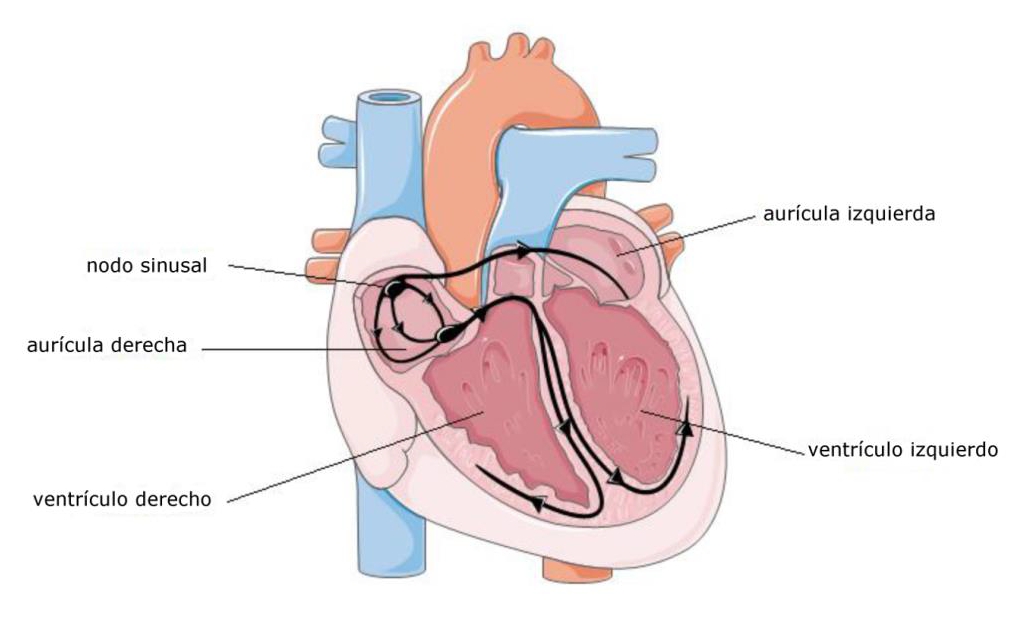
Figura 1: Representación esquemática de la conducción cardíaca
El corazón es un músculo hueco (una bomba) que se compone de cuatro cavidades: dos aurículas (llegada de la sangre), y dos ventrículos (salida de la sangre). El músculo cardíaco (miocardio) tiene la capacidad de contraerse y relajarse según un ritmo muy preciso para que cada cavidad, siguiendo un orden, se llene y seguidamente se vacíe de la sangre que contiene. Esta actividad mecánica está controlada por una actividad eléctrica regular (bajo la forma de "potenciales de acción") relacionada con el paso de iones a través de unas proteínas específicas llamadas canales iónicos presentes en las membranas de las células cardíacas. Esta actividad eléctrica surge en una pequeña zona del miocardio denominada nodo sinusal, situado en la parte alta de la aurícula derecha. A continuación es conducida al resto de células cardíacas por un tejido específico. Esta propagación de la actividad se da primero en las aurículas que se contraen y, después de un pequeño intervalo de tiempo, los ventrículos se contraen a su vez. En todas las células, la contracción es secundaria a la entrada de calcio.
El síndrome de QT largo familiar se produce por mutaciones genéticas, quefundamentalmente se localizan en los genes KCNQ1, KCNH2 y SCN5Q (85%) y tienen como consecuencia anomalías en el funcionamiento de los canales iónicos, dando lugar a un alargamiento del potencial de acción. La anomalía eléctrica puede, en ciertas circunstancias, conducir a alteraciones del ritmo cardíaco (arritmias). Pueden producirse contracciones demasiado rápidas o anárquicas de los ventrículos: taquicardia ventricular, "torsades de pointes" (es una taquicardia ventricular característica conocida con este nombre francés) o fibrilación ventricular. El factor desencadenante (estrés, esfuerzo físico, ralentización del ritmo cardíaco, etc.) varía según el gen, y si la persona sobrecarga su corazón (esfuerzo físico importante, estrés,) puede entonces sufrir un síncope (desmayo) o incluso una parada cardíaca.
¿Cuáles son sus manifestaciones?
Determinadas personas no presentan ninguna manifestación del síndrome (forma asintomática) y es gracias a la realización de un electrocardiograma (ECG) rutinario o en el marco de un estudio sobre la familia que el médico puede observar la anomalía característica del síndrome en el ECG: el alargamiento del intervalo QT (ver figuras 2 y 3).

Figura 2: El electrocardiograma (ECG)
A- Ejemplo de un electrocardiograma normal trazado en papel
https://upload.wikimedia.org/wikipedia/commons/b/bd/12leadECG.jpg
B- El trazo esquemático presente corresponde a la progresión normal de la señal eléctrica durante un latido cardíaco. Ciertas fases (se habla de ondas) tienen una forma característica y corresponden a acontecimientos precisos: onda P = contracción o activación eléctrica (despolarización) de las aurículas, ondas QRS = contracción o activación eléctrica de los ventrículos, onda T= relajación de los ventrículos.
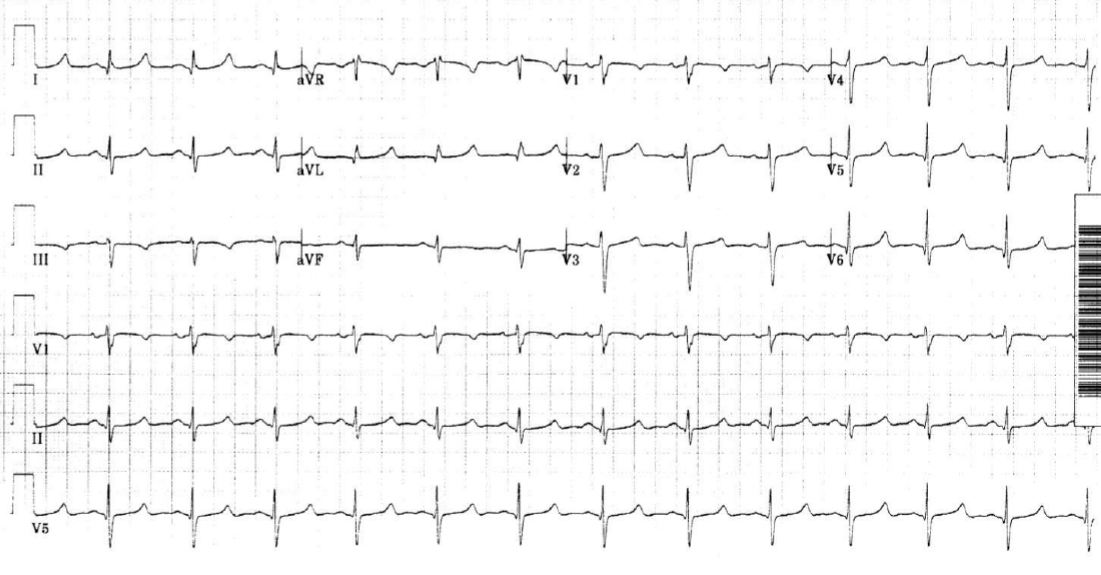
Figura 3: Electrocardiograma anormal con un QT largo (por gentileza del Prof. Hervé Le Marec. Todos los derechos reservados).
En determinadas personas, el síndrome de QT largo se puede manifestar por síncopes (desmayos) desencadenados por esfuerzos físicos (ejercicio, natación, etc.). En otras, estos síncopes pueden producirse ante ruidos intensos o emociones fuertes o sólo se manifiesta si toman determinados medicamentos (algunos antihistamínicos, algunos antibióticos, etc.). A algunas personas se las diagnostica por sufrir convulsiones o pérdidas de orina nocturnas. Las mujeres con el síndrome, tienen un riesgo más elevado de sufrir desmayos durante el período que sigue al parto (post-parto) y en la menopausia. Este riesgo es también más elevado en los niños durante la adolescencia.
En ciertos casos, el síndrome puede provocar una parada cardíaca inesperada y prolongada que, en ausencia de primeros auxilios (masaje cardíaco y/o desfibrilación por descarga eléctrica externa), puede conducir a la muerte (muerte súbita). Desafortunadamente, es la muerte súbita de un joven la que en ocasiones permite realizar el diagnóstico en una familia.
Síndrome de Jervell y Lange-Nielsen
Las personas afectadas de esta forma muy poco frecuente del síndrome de QT largo familiar padecen sordera de nacimiento. Ésta es profunda, es decir que no oyen, ni siquiera sonidos muy fuertes (ver "¿Qué situaciones de discapacidad pueden derivarse de las manifestaciones del síndrome?"). En los recién nacidos afectados, el riesgo de muerte súbita es muy elevado. Sin embargo, actualmente, gracias a la búsqueda sistemática de patologías cardíacas en niños con sordera de nacimiento, puede hacerse un tratamiento precoz.
Síndrome de Timothy
En esta forma excepcional, el riesgo de muerte súbita puede ser elevado. Las personas afectadas presentan también particularidades físicas desde el nacimiento: algunos dedos están mal separados unos de otros, pudiendo estar palmeados o soldados (sindactilia); el tabique nasal está aplanado, las orejas son de implantación más baja de lo normal, la mandíbula y el labio superior son pequeños.
También son posibles signos de autismo.
¿Cuál es su evolución?
La evolución es imprevisible ya que es difícil de prever el riesgo de que se produzcan alteraciones del ritmo cardíaco (taquicardias ventriculares, torsades de pointes).
Por lo general, los desmayos o síncopes y alteraciones del ritmo cardíaco aparecen en la infancia (edad media de 7-8 años). En las mujeres pueden producirse durante toda la vida, y en los varones estos episodios tienden a desaparecer en la edad adulta.
Los síncopes pueden aparecer durante los primeros meses de vida en las formas más raras del SQTL (Jervel y Lange-Nielsen y Timothy) y menos frecuentemente en las formas más comunes del síndrome (Romano-Ward)
En las personas que no saben que tienen el síndrome de QT largo y no están tratadas, el riesgo de síncope y de parada cardíaca es importante. En la gran mayoría de los afectados, los tratamientos permiten reducir este riesgo (ver ¿Existe un tratamiento para este síndrome?).
¿Qué situaciones de discapacidad pueden derivarse de las manifestaciones del síndrome?
Con el fin de evitar todo riesgo de parada cardiaca o de muerte súbita, las personasafectadas deben limitar ciertas actividades físicas.
El ruido puede ser muy molesto para ellas y toda agresión sonora (timbres o ruidosrepentinos) o emocional (positiva o negativa) puede provocar malestar.
El tratamiento con betabloqueantes puede provocar un cansancio importante conconsecuencias en el ámbito escolar, profesional y deportivo. En ocasiones, estosbetabloqueantes pueden provocar sensaciones de frío intenso muy molestas conproblemas de origen circulatorio que afectan a los dedos, la nariz y las orejas(acrosíndrome).
Todas estas situaciones afectan igualmente a las personas que tienen unDesfibrilador Automático Implantado (DAI, ver ¿Existe un tratamiento para este síndrome?)
Para las personas afectadas por el síndrome de Jervell y Lange-Nielsen, la sordera profunda es la causa de una discapacidad que afecta a la comunicación. En la infancia temprana, la deficiencia auditiva representa una limitación para la adquisición del lenguaje y para el aprendizaje de la lectura, la escritura, el cálculo...
Más tarde, la discapacidad puede generar problemas para comunicarse y adquirir nuevas competencias. Un manejo médico adaptado limita las situaciones de discapacidad; sobre todo la implantación coclear precoz permite en los niños adquirir un lenguaje adecuado (ver ¿Cuáles son las ayudas disponibles para prevenir y limitar las situaciones de discapacidad?).
Para las personas afectadas del síndrome de Timothy, la sindactilia de los dedos puede limitar la ejecución de ciertos gestos al dificultar la capacidad de prensión.
El diagnóstico
¿Cuál es su evolución?
Para diagnosticar el síndrome de QT largo, el médico o el cardiólogo se basará en la entrevista, el electrocardiograma (ECG), un holter ECG, incluso la ecografía cardíaca (ver más abajo). El médico podrá asimismo confirmar el diagnóstico y concretar de forma específica el síndrome, solicitando a los pacientes la realización de un test genético en un centro de referencia. Para las personas que no tienen manifestaciones (sin síncope), el diagnóstico se suele hacer por azar durante un ECG rutinario o debido a un estudio familiar tras el diagnóstico de la enfermedad en un miembro de la familia. Ante un desmayo (síncope) en una persona joven, debe realizarse un electrocardiograma (ECG) para descartar la presencia del síndrome.
La entrevista
- Buscar:
- la existencia de episodios de síncopes (desmayos) y averiguar las circunstancias en las que se producen.
- si en la familia algunas personas sufren anomalías del ritmo cardíaco o han sido víctimas de una parada cardíaca inexplicada o de una muerte súbita.
El electrocardiograma (ECG)
El electrocardiograma es el registro de la actividad eléctrica del corazón. Permite confirmar el diagnóstico de QT largo y detectar anomalías de la onda T que pueden sugerir cuál es el gen mutado. El examen es rápido, indoloro, mide el intervalo QT y QTc (QT corregido en función de la frecuencia cardíaca). Para realizarlo, se colocan pequeños sensores de corriente (electrodos) en el tórax, los puños y los tobillos. Un aparato registra la actividad eléctrica del corazón durante algunos minutos. El resultado se presenta en forma de una gráfica (ver figura 2) que será interpretada por el médico.
En el caso del síndrome de QT largo congénito, el ECG muestra de manera característica un alargamiento del tiempo que transcurre entre el inicio de la onda Q y el final de la onda T (ver figura 3). En ocasiones, la forma de la onda T puede indicar cuál es el gen que puede estar mutado (y así diferenciar por ejemplo el SQTL tipo 1, 2 o 3). El trazado del ECG es a veces difícil de interpretar y el médico podrá solicitar la opinión de un cardiólogo experto en el estudio de alteraciones del ritmo cardíaco.
Será necesario realizar un ECG anual.
El Holter ECG
Habitualmente, el médico solicita un registro del ritmo cardíaco durante 24 horas (Holter) que consiste en llevar unos electrodos conectados a un dispositivo que registra regularmente el ritmo cardíaco durante las actividades diarias; a continuación, el registro será analizado por el médico.
En ocasiones, es necesario realizar el ECG durante un esfuerzo (ECG de esfuerzo), sobre todo si el ECG clásico en reposo no permite evidenciar el alargamiento del intervalo QT o las deformaciones de la onda T. El alargamiento del intervalo QT con el esfuerzo nos dará el diagnóstico del síndrome.
La ecografía cardíaca (ecocardiografía)
Este examen indoloro realizado con la ayuda de ultrasonidos, permite asegurar que no hay ninguna anomalía de la estructura del corazón. Para las personas afectadas del síndrome de QT largo, la ecocardiografía no revela ninguna anomalía de la estructura del corazón. Por lo tanto, el examen no es útil para el diagnóstico de QT largo pero sí para descartar otras anomalías en la estructura del corazón.
El test genético (diagnóstico molecular)
El test genético es importante para confirmar el diagnóstico y determinar el gen implicado en el síndrome de QT largo. Consiste en buscar, a partir de una muestra de sangre o saliva, tras la extracción del ADN, la mutación genética causante de la enfermedad. La mutación se identifica en dos de cada tres casos. Los resultados del análisis genético son importantes ya que pueden dar información del riesgo y permiten buscar la presencia de esta anomalía en los otros miembros de la familia.
Este examen debe ser prescrito por un médico con la experiencia suficiente o en un centro de referencia.
¿Es posible confundir esta enfermedad con otras? ¿Cuáles?
Numerosas enfermedades pueden ir acompañadas de desmayos reiterados, como los problemas de tensión arterial (hipotensión ortostática o tensión baja), la bajada de los niveles de azúcar en sangre (hipoglucemia), la actividad excesiva de una parte del sistema nervioso (síncope vasovagal), la epilepsia, y un QT largo causado por un tratamiento con fármacos (QT largo iatrogénico o adquirido).
Pero las circunstancias en las que se producen los desmayos y, sobre todo, el electrocardiograma que debe realizarse ante todo síncope de causa desconocida, nos dará el diagnóstico.
El patrón del ECG característico del síndrome de QT largo permitirá también diferenciarlo de otras anomalías hereditarias del ritmo cardíaco (taquicardia ventricular catecolaminérgica polimórfica, síndrome de Brugada, miocardiopatía hipertrófica, etc.¿).
¿Se puede detectar este síndrome antes de que se manifieste?
Sí. Es posible en el marco de un estudio familiar.(diagnóstico presintomático o predictivo), cuando se conoce un caso en la familia. Una vez que se diagnostica un caso es necesario el estudio de los familiares del mismo. Se hará una entrevista, un ECG y un estudio genético de la mutación identificada en el afectado. Este diagnóstico presintomático permite el manejo de las personas que no tienen todavía manifestaciones del síndrome y evitar el posible riesgo de muerte súbita (ver "¿Existe un tratamiento para este síndrome?").
¿Se puede detectar este síndrome antes de que se manifieste?
El síndrome de QT largo familiar es una enfermedad hereditaria que se transmite de generación en generación, en la mayoría de los casos, de manera "autosómica dominante" (ver figura 4).
El término "autosómica" significa que el gen implicado en la enfermedad no está situado en uno de los cromosomas sexuales (X o Y), sino en uno de los otros 22 pares de cromosomas, los ¿autosomas¿. La enfermedad puede así aparecer en ambos sexos.
Cada individuo posee dos copias de cada gen en un autosoma: una copia se hereda de la madre y la otra del padre.
El término "dominante" significa que es suficiente que una sola copia del gen esté alterada para que la enfermedad se manifieste.
Cuando uno de los padres está afectado, el riesgo de transmisión a la descendencia es del 50%, cualquiera que sea el sexo.
Para un adulto afectado por el síndrome de QT largo (formas dominantes de la enfermedad), el riesgo de transmitir la enfermedad a su hijo es del 50% en cada embarazo.
En menos del 1% de los casos (síndrome de Jervel y Lange-Nielsen), la mutación ha sido transmitida por los dos padres: la transmisión de la enfermedad se produce de manera ¿autosómica recesiva¿ (ver figura 4).
El término ¿recesiva¿ significa que las dos copias del gen deben estar alteradas para que la enfermedad se manifieste.
El término "recesiva" significa que las dos copias del gen deben estar alteradas para que la enfermedad se manifieste.
Si un niño está afectado por el síndrome de Jervell, esto significa que cada uno de sus padres le ha transmitido una copia alterada del gen implicado en el síndrome (el niño es homocigoto para esa mutación). Cada uno de los padres tiene una copia del gen mutado y otra del gen normal; no están enfermos; son heterocigotos. En cada embarazo, el riesgo para la pareja de tener un hijo afectado por el síndrome de Jervell es de uno sobre cuatro (25%), mientras que hay un 50% de probabilidad de tener un hijo con una de las dos mutaciones parentales.
En un adulto afectado por el síndrome de Jervell y Lange-Nielsen, el riesgo de transmitir la enfermedad a su hijo en cada embarazo es muy pequeño, excepto si el otro progenitor es de la misma familia (unión consanguínea) o es portador de una mutación (el riesgo de ser portador de una mutación es el de la frecuencia de los heterocigotos en la población general). Por el contrario, transmitirá obligatoriamente un ejemplar del gen mutado.

Figura 4: Ilustración de las transmisiones autosómica dominante y recesiva
Transmisión autosómica dominante (esquema de la izquierda)
Uno de los padres posee una copia mutada del gen ¿A¿ y está afectado por la enfermedad, como su hijo ¿A/a¿. En cada embarazo, el riesgo de que el hijo de una persona ¿A/a¿ esté enfermo es del 50%. Los hijos ¿a/a¿ no están enfermos y no pueden transmitir la enfermedad (tienen dos copias normales del gen).
Transmisión autosómica recesiva (esquema de la derecha)
Los dos padres son portadores del gen mutado (¿a¿), pero no están enfermos. El hijo ¿a/a¿ ha recibido uno de los dos genes mutados de su padre y el otro de su madre: está afectado por la enfermedad. En cada embarazo, el riesgo de que un hijo de dos personas ¿A/a¿ esté enfermo es del 25%. Los hijos ¿A/a¿ no están enfermos pero son portadores del gen mutado y corren el riesgo de transmitirlo a su descendencia.
El hijo "A/A" no ha heredado ningún gen mutado: no está enfermo y no corre el riesgo de transmitir la enfermedad.
@Orphanet
En alrededor del 15% de los casos, la mutación que causa la enfermedad no se encuentra en ninguno de los progenitores de un niño afectado por el síndrome de Romano-Ward o de Timothy, o solamente en uno de los padres, mientras que sí que se espera que la mutación esté presente en los dos progenitores de un niño afectado por el síndrome de Jervell y Lange-Nielsen. En estos casos, se produce una nueva mutación (mutación "de novo") en el trascurso de la formación de las células reproductoras de los padres. El niño afectado podrá transmitir a su vez el gen mutado a sus hijos según uno de los modos de transmisión descritos anteriormente.
¿Cuáles son los riesgos para otros miembros de la familia?
Los hermanos de una persona afectada tienen un riesgo diferente de padecer la enfermedad según el modo de transmisión del síndrome en la familia.
Para los parentescos de 1er grado (padres, hermanos, hijos), es necesario examinar el árbol genealógico con un genetista o cardiólogo experto, así como una evaluación cardíaca.
El riesgo es de 1:2 en las familias que tienen una enfermedad de transmisión autosómica dominante (la mayor parte de los casos).
En las familias que tienen una enfermedad de transmisión autosómica recesiva (síndrome de Jervell y Lange-Nielsen), el riesgo de transmisión es de 1:4. Sin embargo, esta forma que asocia QT largo y sordera, aparece muy pronto tras el nacimiento, así que si los hermanos mayores de un niño afectado no tienen ninguna manifestación de la enfermedad, en particular si oyen de forma normal (y tienen un ECG normal), tienen un riesgo muy bajo de desarrollar más tarde la enfermedad. Por el contrario, pueden ser portadores de la anomalía genética en el estado heterocigoto y transmitirla a su descendencia.
¿Se puede hacer un Diagnóstico Prenatal (DPN)?
El objetivo del diagnóstico prenatal (DPN) es determinar, durante el embarazo, si el hijo que va a nacer es portador o no de la enfermedad. Para una pareja que ya tiene un hijo afectado o para una persona afectada que desea tener un hijo, es técnicamente posible realizar un DPN si se conoce la mutación responsable de la enfermedad. Sin embargo, esta opción solamente se propone si la enfermedad es de una gravedad considerable y reconocida como incurable. En la práctica, afecta casi únicamente a las familias afectadas por el síndrome de Jervell y Lange-Nielsen.
La realización del diagnóstico prenatal debe discutirse antes con los genetistas y en consulta de diagnóstico prenatal, sobre todo para evaluar la gravedad de la enfermedad ya que los exámenes necesarios conllevan un riesgo de aborto espontáneo (de 0,5 al 1-2%).
El tratamiento, la atención, la prevención
¿Existe un tratamiento para este síndrome?
No es posible curar el síndrome de QT largo. Por el contrario, la prevención y los tratamientos permiten limitar considerablemente el riesgo de síncopes y/o de paradas cardíacas. Con el fin de prevenir este riesgo, se aconseja administrar un tratamiento a la mayoría de las personas diagnosticadas. Éstas deberían llevar consigo la lista de medicamentos contraindicados que prolongan el QT (ver anexo de la ficha Orphanet Urgencias sobre el síndrome de QT largo) y respetar las restricciones sobre la actividad deportiva.
Los medicamentos
Los objetivos del tratamiento betabloqueante en pacientes asintomáticos (salvo contraindicaciones específicas) son reducir los riesgos de síncopes y de muerte súbita. Por lo general, el médico prescribe un betabloqueante (por ejemplo, el nadolol), a excepción del sotalol, que está contraindicado. Los betabloqueantes son una familia de medicamentos que actúan sobre el ritmo cardíaco ¿ son antiarrítmicos ¿ y son los únicos medicamentos que pueden reducir el riesgo de muerte súbita que existe durante el síndrome de QT largo. La dosis adecuada a tomar se determinará para cada persona teniendo en cuenta sus características (edad, sexo, perfil genético y aspecto del ECG y del Holter-ECG). La eficacia del tratamiento y la aparición de efectos secundarios serán evaluados y la dosis podrá ajustarse si es necesario.
Desfibrilador automático implantable
Si los síncopes se repiten, o si los betabloqueantes están contraindicados y el riesgo de sufrir una arritmia es elevado, se implantará un desfibrilador automático (Desfibrilador Automático Implantable ¿ DAI ¿ o Desfibrilador Cardioversor Implantable ¿ DCI). El desfibrilador controla permanentemente el corazón y cuando detecta una alteración del ritmo, libera una descarga eléctrica con el fin de corregiresa alteración. El dispositivo se coloca, con anestesia local, bajo la piel, cerca de la clavícula y está unido al corazón por una sonda introducida por una vena en el hueco del hombro.
La persona que porta un DAI debe asegurarse siempre de tener consigo la tarjeta de identificación del fabricante y el modelo del dispositivo (tarjeta europea de portador de DAI). Debe tener un seguimiento cardiológico regular, al menos anualmente y, obligatoriamente consultar de urgencia a un cardiólogo en caso de descarga del dispositivo.
El DAI puede implantarse a cualquier edad y en niños pequeños existen técnicas de implantación abdominal debido al voluminoso tamaño del dispositivo, pero sin embargo esta indicación es excepcional.
El portador de un DAI tiene que consultar antes de realizarse una Imagen por Resonancia Magnética (RM), ya que hay algunos modelos de DAI que son incompatibles con las resonancia.
Simpatectomía izquierda
En algunos casos muy poco frecuentes, puede sugerirse una intervención quirúrgica: la denervación simpática cardíaca izquierda ¿ o simpatectomía izquierda, en caso de síncopes de repetición en pacientes que ya están tratados con DAI y betabloqueantes mientras que la persona recibe un tratamiento con betabloqueantes y porta un DAI. Se trata de extraer los ganglios nerviosos (o ganglios simpáticos) a nivel del tórax.
¿Cuáles son las ayudas disponibles para prevenir y limitar las situaciones de discapacidad?
La mayoría de personas afectadas por el síndrome tienen una vida diaria normal que no se ve limitada por la enfermedad excepto en los esfuerzos físicos intensos y el deporte (ver más abajo).
En los niños afectados por el síndrome de Jervell y Lange-Nielson, la sordera causa una deficiencia que debe compensarse rápidamente para que puedan adquirir el lenguaje. Actualmente, los niños pueden beneficiarse de unos implantes cocleares que se colocan mediante un procedimiento quirúrgico. Estos restablecen una audición diferente de la normal y necesitan una reeducación auditiva (ortofonía), pero por lo general permitirán la adquisición de un lenguaje de buena calidad. Los sistemas de ayuda para la comprensión de mensajes sonoros (inducción magnética, bluetooth u ondas FM) son compatibles con el DAI.
Para los niños afectados por el síndrome de Timothy, la sindactilia puede ser corregida quirúrgicamente.
¿Es recomendable recibir apoyo psicológico?
Las personas afectadas por el síndrome de QT largo congénito y su entorno pueden necesitar apoyo psicológico, por ejemplo en el momento del diagnóstico, para aceptar este diagnóstico y expresar sus miedos y creencias respeto al riesgo para la vida.
El apoyo psicológico es mucho más útil cuando la forma del síndrome es grave (síndrome de Jervell y Lange-Nielsen y síndrome de Timothy).
¿Qué puede hacer uno mismo para prevenir las manifestaciones del síndrome?
Es fundamental conocer y evitar los medicamentos y las bebidas energéticas que prolongan el QT.
Asimismo es importante llevar consigo la tarjeta europea de portador de DAI y la lista de medicamentos contraindicados (ver anexo de la ficha Orphanet Urgencias sobre el Síndrome de QT largo).
De igual manera, es posible inscribirse gratuitamente en el sitio web Crediblemeds.org (https://www.crediblemeds.org/index.php/login/dlcheck) para estar informados de las actualizaciones en relación a la lista de medicamentos contraindicados.
La práctica de una actividad física regular está recomendada pero sólo se autoriza la participación en deportes recreativos y únicamente si se respeta de manera estricta el tratamiento betabloqueante y éste es eficaz. La práctica de deportes competitivos está totalmente contraindicada, al igual que la de determinados deportes en los que un desmayo podría exponerle a un riesgo mayor (buceo, alpinismo¿) (ver ¿¿Qué consecuencias tiene la enfermedad en la vida diaria¿?): se aconseja tratar este asunto de forma individualizada con el cardiólogo.
La persona afectada deberá asegurarse de mantener un buen aporte de potasio. De hecho, la insuficiencia de potasio (hipopotasemia) favorece las alteraciones del ritmo cardíaco. Toda insuficiencia deberá corregirse con suplementos de potasio.
En caso de implantación de un desfibrilador automático, un programa de educación terapéutica puede permitir al paciente un mejor manejo en el día a día y ocuparse mejor de su DAI según sus hábitos personales, su modo de vida, etc. Por ejemplo, aprender cómo actuar cuando el desfibrilador administra una descarga eléctrica (sentarse o tumbarse, no permanecer solo y consultar al cardiólogo cuando remita para hacer una evaluación...) o saber cuáles son los síntomas de alarma que obligarían a contactar con un médico de emergencias. Las personas que portan un DAI deben tener su tarjeta siempre con ellas (ver más abajo).
En cualquier caso, es importante hacer un seguimiento médico regular; además, en caso de embarazo, las mujeres deben consultar al cardiólogo ya que existen riesgos específicos al final del mismo y durante el post-parto.
¿Cómo realizar el seguimiento?
Las personas afectadas pueden hacerse un seguimiento en las consultas especializadas (centros de referencia) de alteraciones del ritmo cardíaco de origen genético o de enfermedades cardíacas hereditarias o cardiopatías familiares. Los datos de contacto están disponibles en el sitio web de Orphanet (www.orphanet.es). El seguimiento lo realizará un cardiólogo, en colaboración con el médico de cabecera.
El seguimiento regular permite, sobre todo al cardiólogo, evaluar el efecto de los tratamientos y sus posibles efectos secundarios, y si es necesario, confirmar que el DAI es eficaz y bien tolerado. Además, permite verificar con el médico de cabecera que la persona ha entendido bien los elementos a vigilar.
Por regla general, el seguimiento se hace al menos una vez al año e incluye una entrevista y un examen clínico (ritmo cardíaco, presión arterial, peso¿). Además, puede comprender determinados exámenes (ECG, Holter-ECG, pruebas de esfuerzo), permitiendo verificar que la dosificación y la efectividad de los betabloqueantes son satisfactorias.
Todo nuevo síntoma, síncope o desmayo o pérdida de conciencia, debe ser informado al médico junto con la lista de medicamentos contraindicados (ver anexo de la ficha Orphanet Urgencias sobre el Síndrome de QT largo).
¿Qué información debemos conocer y transmitir en caso de emergencia?
De forma general, es importante informar de que se está afectado por el síndrome de QT largo y mencionar el tratamiento que se sigue. Esto permite evitar medicamentos que están contraindicados. Cuando una persona está afectada por el síndrome de QT largo, existen contraindicaciones y precauciones de empleo en numerosos medicamentos.
También es importante señalar que se porta un Desfibrilador Automático Implantable (DAI) y llevar consigo la tarjeta europea de portador de DAI permanente ya que ciertos exámenes podrían estar contraindicados (IRM, por ejemplo).
Con el fin de transmitir toda la información importante en caso de emergencia, se ha elaborado una ficha de Orphanet Urgencias, con la lista de medicamentos contraindicados en el Anexo.
Vivir con: situaciones de discapacidad por el síndrome de QT largo en el día a día
¿Qué consecuencias tiene la enfermedad en la vida diaria?
La mayor parte del tiempo, este síndrome tiene poco impacto en la vida familiar, social y profesional. Sin embargo, algunos padres pueden sentirse abrumados por la afectación de sus hijos y sobreprotegerlos. La única contraindicación es la práctica de una actividad deportiva en competición.
La práctica de una actividad física de ocio, que en ningún caso debe ser intensa, está autorizada si se respeta el tratamiento betabloqueante y la hipopotasemia (insuficiencia en potasio) está corregida. Ciertos deportes están estrictamente prohibidos: deportes acuáticos, baloncesto, fútbol, tenis, rugby, buceo, escalada¿ Es necesario consultar a un cardiólogo antes de comenzar cualquier actividad.
En el caso de portar un DAI, las limitaciones en la vida diaria son más importantes. Se insistirá sobre ello con un programa de educación terapéutica. La conducción automovilística requiere que se adopten ciertas precauciones e incluso puede prohibirse en determinados casos: al menos en las primeras semanas que siguen a la implantación.
Los viajes no están contraindicados. En aeropuertos, al pasar un control de seguridad, habrá que indicar al personal si se es portador de un DAI y mostrar la tarjeta identificativa. El paso a través de un control de seguridad debe hacerse tranquilamente, sin detenerse.
¿Qué consecuencias tiene la enfermedad en la escolaridad y en la práctica deportiva?
La legislación española establece la obligación de las Administraciones educativas de asegurar los recursos necesarios para los alumnos que presentan necesidades educativas especiales. Los mecanismos de refuerzo que deberán ponerse en práctica tan pronto como se detecten dificultades de aprendizaje podrán ser tanto organizativos como curriculares. Entre otras medidas, podrán considerarse el apoyo en el grupo ordinario, los agrupamientos flexibles o las adaptaciones del currículo. Por tanto, el centro escolar deberá poner en marcha un plan de acogida para facilitar la integración y la resolución de las dificultades que pueda encontrar el niño, como el manejo del paro cardíaco y actividad deportiva adaptada. Es especialmente importante que el centro escolar esté informado de la existencia y el uso del desfibrilador externo semiautomático (DESA).
El Servicio de Atención Educativa de FEDER (inclusion@enfermedades-raras.org), le facilitará apoyo y orientación en relación a las necesidades educativas del alumnado afectado por una enfermedad poco frecuente.
El departamento de Educación del Centro de Referencia Estatal de Atención a Personas con Enfermedades Raras y sus Familias (Creer) desarrolla acciones para la inclusión educativa del alumnado con enfermedades raras, dirigido a toda la comunidad educativa. Ofrece servicios de información, orientación y asesoramiento para las familias, alumnado, profesionales y centros escolares a través del e-mail: escuela@creenfermedadesraras.es
¿Qué consecuencias tiene la enfermedad en el embarazo?
En líneas generales, se deberán tomar en cuenta las siguientes recomendaciones y advertencias:
- El tratamiento con betabloqueantes no deberá interrumpirse durante el embarazo ni durante el postparto.
- El tratamiento con betabloqueantes durante la lactancia debe discutirse con el equipo médico caso por caso.
- Existe un riesgo bajo para el recién nacido de sufrir una hipoglucemia o de tener problemas cardíacos al nacer (síndrome de QT largo neonatal).
- Se recomienda que las mujeres embarazadas sean atendidas en una sala de maternidad habilitada para embarazos de riesgo (de nivel 2 mínimo).
- Al nacimiento, se recomienda realizar al niño una evaluación cardiológica (ECG, Holter) y un diagnóstico genético si la mutación familiar es conocida, con el objetivo de tomar medidas de prevención si se confirma el diagnóstico.
¿En qué punto se encuentra la investigación?
Las investigaciones apuntan, en parte, a un mejor análisis de la relación entre las diferentes mutaciones responsables del síndrome y la gravedad de las manifestaciones.
También van dirigidas a desarrollar nuevas herramientas de diagnóstico y posibles tratamientos.
Nuevos Desfibriladores Automáticos Implantables (DAI) sin sonda (sin cables) están en proceso de evaluación.
¿Cómo puedo entrar en contacto con otros afectados por la misma enfermedad?
Contactando con las asociaciones de pacientes dedicadas a esta enfermedad. Pueden obtener sus datos de contacto en la página web de Orphanet (www.orphanet.es) o a través del Servicio de Información y Orientación sobre Enfermedades Raras (SIO) de la Federación Española de Enfermedades Raras (+34 918 221 725, sio@enfermedades-raras.org)
Las prestaciones sociales en España
La legislación española reconoce a las personas con discapacidad una serie de derechos en todos los ámbitos, desde la protección de la salud, a la atención integral, incluida la educación y el empleo. Es, por tanto, una obligación del Estado y de los poderes públicos garantizar la prevención, los cuidados médicos y psicológicos, la rehabilitación adecuada y los recursos económicos para facilitar una mayor realización personal e integración laboral y social a las personas con discapacidad física, intelectual o sensorial, proporcionando la tutela necesaria a las personas que lo necesiten.
La Ley de Integración Social de las personas con discapacidad (Lismi), contempla un conjunto de prestaciones económicas y técnicas (Asistencia Sanitaria y Prestación Farmacéutica (ASPF)) destinadas a la protección de las personas discapacitadas que por no desarrollar actividad laboral no están comprendidas en el campo de aplicación de la Seguridad Social.
La gestión y reconocimiento del derecho a percibir una prestación social y económica la realizan las Comunidades Autónomas, excepto en el caso de Ceuta y Melilla, ciudades en las que el Instituto de Mayores y Servicios Sociales (Imserso) se hace cargo de la gestión directamente. La solicitud podrá presentarse personalmente en las oficinas de los Servicios Sociales de las Comunidades Autónomas, del Imserso o en cualquier otra de la Seguridad Social, en las que se facilitará el impreso correspondiente, o por correo. La información relativa a las direcciones y teléfonos de información de las Comunidades Autónomas y Direcciones Territoriales del Imserso, puede ser consultada en su sitio web.
Dr. Roberto Barriales.
Unidad de Cardiopatías Familiares.
Hospital Universitario de A Coruña